储存条件:短期保存 2~8℃,长期保存 -20 ~ -80℃

天然产物作为药物发现的重要源泉,在人类与疾病的漫长斗争史中扮演着不可替代的角色。从古老的植物药用到现代靶向药物的开发,自然界中结构多样的次生代谢产物始终是创新药物先导化合物的宝库。在众多具有生物活性的天然产物家族中,氧杂蒽酮(Xanthone)类化合物因其独特的化学骨架和广泛的药理活性而备受关注。这类化合物通常存在于龙胆科、藤黄科、豆科等植物中,展现出抗炎、抗氧化、抗肿瘤、抗菌及神经保护等多种生物活性。
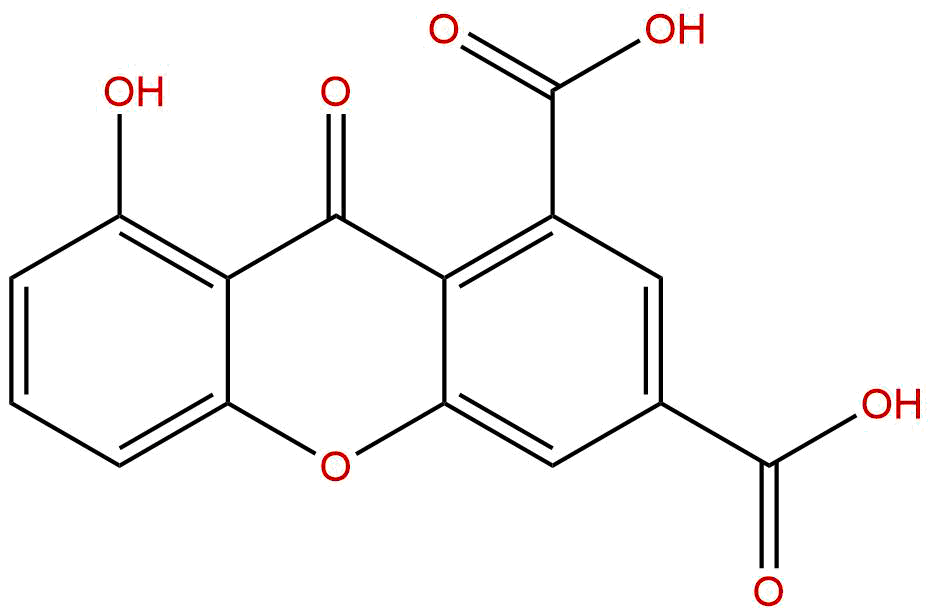
决明山酮(Cassiaxanthone),CAS号为28917-02-4,是一种从豆科植物决明属(Cassia)中分离得到的天然氧杂蒽酮类化合物。其名称直接指向其植物来源——决明属植物。作为该属植物中特征性的活性成分之一,决明山酮近年来逐渐进入研究者的视野。初步研究表明,该化合物可能具有抗炎、抗氧化以及潜在的抗肿瘤活性,但其深入的药理机制、分子靶点以及系统性的成药性评价尚处于起步阶段。鉴于天然产物研究的不断深入以及现代药物研发对新颖化学实体(NCE)的迫切需求,系统梳理决明山酮的研究现状,评估其作为先导化合物的潜力,对于推动其后续开发具有重要意义。本文旨在对决明山酮的化学结构、植物来源、提取方法、药理活性、作用机制、成药性及临床应用前景进行全面而深入的综述,以期为该领域的进一步研究提供参考。
决明山酮属于氧杂蒽酮类化合物,其核心骨架由两个苯环通过一个含氧的六元杂环稠合而成(二苯并-γ-吡喃酮)。这种三环结构赋予了氧杂蒽酮类化合物独特的平面性和芳香性,使其能够与多种生物大分子(如蛋白质、DNA)发生π-π堆积、氢键及疏水相互作用。决明山酮的具体结构通常带有羟基、甲氧基等取代基,这些取代基的种类、数目和位置直接决定了其理化性质和生物活性。根据现有文献,决明山酮的分子式为C₁₆H₁₂O₆,分子量为300.2200 g/mol。其结构中通常包含多个酚羟基,这使得该化合物具有一定的极性和酸性,同时也赋予了其良好的抗氧化能力——酚羟基能够有效清除自由基。
在理化性质方面,基于其分子结构可以推断,决明山酮为黄色或淡黄色结晶性粉末,具有一定的熔点。由于其分子中含有多个极性基团(如-OH),它在极性溶剂(如甲醇、乙醇、二甲基亚砜)中具有较好的溶解度,而在水中的溶解度则相对较低。这种亲脂性与亲水性的平衡(通常以油水分配系数LogP衡量)对其在生物体内的吸收、分布、代谢和排泄(ADME)过程至关重要。初步的成药性参数显示,其分子量(300.22 Da)符合“类药五规则”(Lipinski’s Rule of Five)中分子量小于500的要求,提示其具有较好的口服吸收潜力。然而,关于其血脑屏障透过性、肝毒性、心脏毒性(如hERG通道抑制)以及遗传毒性(Ames试验)等关键数据,目前均标注为“Unknown”,这构成了其成药性评价中的关键空白,也是未来研究需要优先突破的方向。
决明山酮的主要植物来源是豆科(Fabaceae)决明属(Cassia)植物。决明属是一个庞大的植物类群,包含约600种植物,广泛分布于热带和亚热带地区。许多决明属植物在传统医学中有着悠久的应用历史,例如,决明(Cassia obtusifolia L.)的种子被用作清肝明目、润肠通便的药材;番泻叶(Cassia angustifolia Vahl)则以其泻下作用而闻名。决明山酮正是从这些药用植物中分离鉴定出的活性成分之一。除了决明属,有报道称该化合物也可能存在于其他属植物中,但决明属植物仍是其主要且最可靠的来源。
从植物原料中提取决明山酮,通常遵循天然产物化学的经典流程,主要包括以下几个步骤:
尽管决明山酮的研究历史相对较短,但已有的药理学研究揭示了其具有多种生物活性,展现出作为多靶点天然产物的潜力。
1. 抗氧化活性
这是氧杂蒽酮类化合物最普遍且研究最深入的活性之一。决明山酮分子结构中的酚羟基是有效的氢原子供体,能够中和活性氧(ROS)和活性氮(RNS),如羟基自由基(·OH)、超氧阴离子(O₂⁻·)和过氧亚硝酸盐(ONOO⁻)。体外化学实验(如DPPH、ABTS、FRAP法)已证实其具有显著的抗氧化能力。这种抗氧化活性是其发挥其他药理作用(如抗炎、保肝)的重要基础。
2. 抗炎活性
炎症是多种慢性疾病(如心血管疾病、糖尿病、神经退行性疾病)的共同病理基础。研究表明,决明山酮能够抑制脂多糖(LPS)刺激的巨噬细胞(如RAW 264.7细胞)中一氧化氮(NO)和前列腺素E₂(PGE₂)的产生。其机制可能与抑制诱导型一氧化氮合酶(iNOS)和环氧合酶-2(COX-2)的表达有关。此外,它还可能通过抑制核因子-κB(NF-κB)或丝裂原活化蛋白激酶(MAPK)信号通路的激活,从而减少下游促炎细胞因子(如TNF-α、IL-1β、IL-6)的释放。
3. 抗肿瘤活性
初步的细胞实验显示,决明山酮对某些肿瘤细胞株(如肝癌细胞HepG2、乳腺癌细胞MCF-7、结肠癌细胞HT-29)具有增殖抑制作用。其作用机制可能涉及多个方面:诱导细胞周期阻滞(如将细胞阻滞在G0/G1期或G2/M期)、诱导细胞凋亡(通过激活Caspase家族蛋白、调节Bcl-2/Bax比例)、抑制肿瘤细胞迁移和侵袭(可能通过下调基质金属蛋白酶MMP-2/9的表达)。然而,目前的研究多停留在体外细胞水平,缺乏体内动物模型的验证,其抗肿瘤谱和选择性也有待进一步明确。
4. 抗菌活性
一些研究报道了决明山酮对某些细菌和真菌具有一定的抑制作用。例如,它可能对金黄色葡萄球菌(Staphylococcus aureus)、枯草芽孢杆菌(Bacillus subtilis)等革兰氏阳性菌表现出抑制活性,但对革兰氏阴性菌的活性通常较弱。其抗菌机制可能与破坏细菌细胞膜完整性或抑制细菌关键酶活性有关。
5. 其他活性
此外,还有零星报道指出决明山酮可能具有保肝、降血糖或神经保护等活性。例如,在肝细胞损伤模型中,它可能通过抗氧化和抗炎作用减轻肝损伤;在糖尿病模型中,它可能通过抑制α-葡萄糖苷酶活性来延缓碳水化合物的吸收。但这些发现尚需更多、更系统的研究来证实。
深入理解决明山酮的作用机制和分子靶点,是将其从天然产物转化为先导化合物的关键。目前,关于其分子机制的研究尚不深入,但基于其结构特征和有限的药理数据,可以推测其可能的作用模式。
1. 信号通路调控
- NF-κB通路:NF-κB是炎症和免疫反应的核心转录因子。在静息状态下,NF-κB与IκB蛋白结合,以无活性形式存在于细胞质中。当受到LPS、TNF-α等刺激时,IκB激酶(IKK)被激活,导致IκB磷酸化并降解,释放NF-κB进入细胞核,启动靶基因(如iNOS, COX-2, TNF-α)的转录。决明山酮可能通过抑制IKK的活性或阻止IκB的降解,从而阻断NF-κB的核转位,发挥抗炎作用。
- MAPK通路:MAPK家族包括ERK、JNK和p38三条主要通路,参与调控细胞增殖、分化、凋亡和炎症反应。研究表明,某些氧杂蒽酮类化合物能够抑制LPS诱导的p38和JNK的磷酸化,从而减少炎症介质的产生。决明山酮可能具有类似的作用。
- PI3K/Akt/mTOR通路:该通路是调控细胞生长、增殖和存活的关键信号通路,在多种癌症中异常激活。决明山酮可能通过抑制PI3K或Akt的磷酸化,进而抑制mTOR的活性,诱导肿瘤细胞自噬或凋亡。
2. 直接分子靶点
- 酶抑制:基于其平面芳香环结构,决明山酮可能作为某些酶的抑制剂。例如,它可能直接与iNOS、COX-2、α-葡萄糖苷酶或拓扑异构酶等靶点结合,抑制其催化活性。分子对接(Molecular Docking)技术是预测其与潜在靶点结合模式的有力工具,但需要后续的酶学实验(如表面等离子体共振SPR、等温滴定量热ITC)进行验证。
- 受体调节:虽然报道较少,但也不排除决明山酮与某些细胞表面受体(如Toll样受体TLR4、雌激素受体ER)或核受体(如过氧化物酶体增殖物激活受体PPARγ)相互作用,从而调节下游信号。
3. 表观遗传调控
近年来,天然产物对表观遗传修饰的影响日益受到关注。决明山酮是否能够通过抑制组蛋白去乙酰化酶(HDAC)或DNA甲基转移酶(DNMT)的活性,来改变染色质状态和基因表达,是一个值得探索的新方向。
总体而言,决明山酮的作用机制呈现出“多靶点、多通路”的特征,这既是天然产物的优势(可能产生协同效应,降低耐药性),也为其机制研究的深入带来了挑战。未来需要结合转录组学、蛋白质组学等系统生物学方法,结合基因敲除/敲入等分子生物学技术,来精确描绘其作用网络和关键靶点。
一个化合物要成为候选药物,不仅需要具有优良的药理活性,还必须具备良好的成药性(Drug-likeness)和可接受的药代动力学(ADME)特性。对于决明山酮而言,这方面的研究是其从“活性化合物”迈向“先导化合物”的最大瓶颈。
1. 类药性分析
根据Lipinski五规则,决明山酮的分子量(300.22 Da < 500)、氢键供体数(酚羟基,通常>5?需具体结构确认,但一般氧杂蒽酮类化合物氢键供体数在3-5之间)、氢键受体数(氧原子,通常<10)以及脂水分配系数(LogP,需实验测定,预测值可能在2-4之间)均可能在可接受范围内。这表明其理论上具有较好的口服生物利用度潜力。然而,五规则只是一个初步筛选标准,不能完全代表真实的体内行为。
2. 关键ADME特性
- 吸收:决明山酮的水溶性较差,这可能是限制其口服吸收的主要因素。其渗透性如何?是主动转运还是被动扩散?是否P-糖蛋白(P-gp)的底物?这些问题均不清楚。
- 分布:其血浆蛋白结合率、表观分布容积(Vd)未知。特别是血脑屏障(BBB)透过性标注为“Unknown”,这对于评估其在中枢神经系统疾病(如神经退行性疾病)中的应用潜力至关重要。
- 代谢:肝脏是药物代谢的主要器官。决明山酮的酚羟基是I相代谢(如葡萄糖醛酸化、硫酸化)和II相代谢(如甲基化)的潜在位点。其是否会被细胞色素P450酶系(CYP450)代谢?代谢产物是否具有活性或毒性?这些都是未知数。肝毒性标注为“Unknown”,提示其肝脏安全性尚待评估。
- 排泄:决明山酮及其代谢产物主要通过胆汁还是肾脏排泄?其半衰期(t₁/₂)是多少?这些数据完全空白。
3. 安全性评价
- 心脏毒性:hERG(human Ether-à-go-go Related Gene)钾离子通道抑制是导致药物性QT间期延长和致命性心律失常(尖端扭转型室性心动过速)的主要原因。决明山酮对hERG通道的抑制活性为“Unknown”,这是其安全性评价中的高风险项,必须优先进行体外hERG膜片钳实验进行评估。
- 遗传毒性:Ames试验是检测化合物致突变性的标准方法。结果为“Unknown”,意味着其潜在的遗传毒性风险未知。如果该化合物表现出抗肿瘤活性,其可能通过DNA损伤机制发挥作用,但这也会带来致癌风险。因此,Ames试验是必须进行的。
- 其他毒性:急性毒性、亚慢性毒性、生殖毒性等均未开展。
4. 结构修饰与优化
鉴于决明山酮在溶解性、代谢稳定性及潜在毒性方面可能存在的问题,药物化学家可以通过结构修饰来改善其成药性。例如:
- 引入亲水基团:在适当位置引入氨基、羧基或糖基,提高水溶性。
- 屏蔽代谢位点:将易代谢的酚羟基进行甲基化或乙基化,提高代谢稳定性。
- 前药设计:将酚羟基制成磷酸酯或氨基酸酯前药,改善水溶性和口服吸收,在体内经酶解或水解后释放原药。
尽管决明山酮的研究仍处于早期阶段,但其独特的化学结构和初步发现的药理活性,为其在多个疾病领域的应用前景提供了想象空间。
1. 炎症性疾病
基于其抗炎活性,决明山酮或其衍生物有潜力被开发为治疗慢性炎症性疾病的药物,如类风湿性关节炎、炎症性肠病(IBD)或哮喘。其多靶点的抗炎机制可能比单一靶点的非甾体抗炎药(NSAIDs)具有更低的胃肠道副作用。
2. 肿瘤辅助治疗
虽然其直接的抗肿瘤活性可能不够强,但决明山酮可以作为化疗增敏剂或放疗增敏剂,与现有化疗药物(如顺铂、阿霉素)联合使用,以降低化疗药物的剂量和毒副作用,或逆转肿瘤的多药耐药性(MDR)。此外,其抗氧化活性也使其有潜力作为癌症预防剂。
3. 代谢性疾病
其对α-葡萄糖苷酶的抑制活性,提示其可能用于2型糖尿病的治疗。此外,其抗氧化和抗炎活性也可能对糖尿病并发症(如肾病、视网膜病变)具有保护作用。
4. 神经退行性疾病
如果能够解决其BBB透过性问题,决明山酮的抗氧化和抗炎特性使其在阿尔茨海默病(AD)和帕金森病(PD)的治疗中具有潜在价值。它可能通过抑制β-淀粉样蛋白(Aβ)聚集、清除自由基、抑制神经炎症等途径发挥神经保护作用。
未来研究方向与挑战:
决明山酮作为豆科决明属植物中一种重要的天然氧杂蒽酮类化合物,凭借其独特的化学结构和初步揭示的抗氧化、抗炎、抗肿瘤等多重药理活性,展现了作为药物先导化合物的潜力。然而,目前的研究现状犹如冰山一角,其深入的药理机制、确切的分子靶点以及至关重要的成药性评价(尤其是ADME/Tox特性)均存在大量空白。从“天然产物”到“创新药物”的转化之路,充满了机遇,更伴随着巨大的挑战。未来的研究必须将重点从简单的活性筛选转向系统性的成药性评价和深入的机制探索。只有通过药物化学、药理学、药代动力学和毒理学等多学科的协同攻关,才能科学地评估决明山酮的真正价值,并最终决定其能否从实验室走向临床,为人类健康事业做出贡献。在天然药物化学这片广袤的星空中,决明山酮这颗“明星”能否真正闪耀,尚需时间的检验和研究者们的不懈努力。

版权所有:© 成都普瑞法科技开发有限公司(2015)备案号:蜀ICP备15035167号-1 客服热线:400-829-7929
技术支持:南京库价